5. Initial analysis with Sequedex: Phylogenetic and functional profiles¶
As described in How is Sequedex used with other software?, Sequedex can be applied to a wide variety of problems. While it is possible that a cursory examination of Sequedex output will be sufficient to address the user’s question to the appropriate level of confidence, we expect most Sequedex users will need to combine their Sequedex analysis with other techniques and software packages. We provide examples of a variety of these analyses, including:
- the direct examination of phylogenetic or functional profiles with :program: Excel, described in this chapter.
- the visualization and characterization of these profiles, described in Using Sequestat
- the analysis of the individual reads retrieved, described in Annotated reads, and
- the multi-sample comparisons, described in Comparing Samples.
In these chapters, we will draw upon software tools and techniques that the user may not be familiar with or may not have installed on their system. To help these users, we provide Additional software tools and resources for use in conjunction with Sequedex.
It is assumed at this point that the user is comfortable running Sequescan and can obtain multiple output files for comparison. Please refer to the previous two chapters for installation, platform-dependent instructions on running Sequescan with both the GUI and the command line, and instructions on obtaining a license.
The ability of Sequedex to profile bacterial communities with metagenomic data leads naturally to the question of how a user’s microbial community compares to previously sequenced microbial communities. In this chapter, we lead the user through the process of computing phylogenetic profiles and comparing them to publically available datasets. In additional to phylogenetic information, the sequences obtained in a shotgun metagenomics dataset can be used to obtain functional profiles of genes in a microbial community, identify the presence of particular genes of interest, or even determine which proteins are the most important determinants of classification within a set of microbial communities. In this chapter, we we lead the user through the process of computing functional profiles.
A note of caution is warranted here. In this and the next section we present phylogenetic and functional profiles, as well as analysis of these profiles across an extraordinary range of microbial communities with samples prepared by different researchers with a variety of purposes, sequenced by different methods with different read-lengths. We have primarily relied on publically available datasets with associated publications, although a few examples of unpublished work with experimental collaborators is also included. Our purpose in this user’s manual is to provide illustrative examples of how phylogenetic and functional profiles can be visualized and analyzed for both self-consistency and biological insights, and provide analysis of a reference collection of data to which the user can compare his or her own data. The job of cross-checking these insights with further experimentation analysis is distinct and belongs in the peer-reviewed literature. Our intent here is to put this analysis tool into the hands of the people best-suited to perform these cross-checks - those who decide which samples will be prepared and know what biological phenomonena are expected.
5.1. Acquiring data files¶
Sequedex at present requires input sequence data files be in fasta, fastq, or gzipped fasta or fastq data format. Sequencing platforms typically output sequences in a format with quality scores, such as fastq, which can be used as input to assemblers. Because possibility space of 10-mers of amino acids is so much larger than the number of signatures, Sequedex is relatively insensitive to sequencing errors and the quality scores are ignored, and the fasta file format is suitable. If the user’s data is in another format, the it must be changed into fasta or fastq format. While preprocessing of reads to remove duplicates is valuable, and minimal quality score filtering is probably useful, assembling the data into contigs before profiling the community is probably unhelpful, as it makes the profiles dependent on the depth of sequencing.
For users wishing to compare their data to others, metagenomics data can be acquired from several sources, including the Sequence Read Archive at NCBI, where the production phase of the human microbiome project can be found with the project number, SRP002163 and the study number SRP002163. Individual data sets can be downloaded from their ftp site with a web browser (or using wget) at ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR059/SRR059366/SRR059366.sra. Metadata about data sets, such as it is, can be obtained with the Run Browser at the Sequence Read Archive, http://www.ncbi.nlm.nih.gov/Traces/sra/?view=run_browser/. The human microbiome data is probably easier to obtain from the Human Microbiome Project website: http://www.hmpdacc.org.
Environmental metagenomics data sets can be found at NCBI with the taxonid 410658. A wide variety of individual projects can be found, including samples from Pru Toh Daeng Peat Swamp, in Southern Thailand: SRR023820, which can be downloaded from ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP001/SRP001114/SRR023820/SRR023820.sra.
In order to process data sets from the Sequence Read Archive, it is necessary to extract fasta or fastq files from the .sra files obtained above. This can be done with the fastq-dump utility from the SRA Toolkit, available for Linux, Mac, and Windows platforms at http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software. Generation of a fastq or [fasta] file from the HMB dataset above requires the command:
fastq-dump [--fasta] SRR059366.sra
The Department of Energy’s Joint Genome institute also maintains a repository of metagenomic sequence data at their integrated metagenomics (IM) site.
The CREO data from reference [1] can be downloaded in fasta format at http://img.jgi.doe.gov/cgi-bin/m/main.cgi?section=MetaDetail&downloadTaxonReadsFnaFile=1&taxon_oid=2029527002&noHeader=1, while additional metagenomics datasets are listed at http://img.jgi.doe.gov/cgi-bin/m/main.cgi?section=TaxonList&page=taxonListAlpha&domain=*Microbiome.
The Community Cyberinfrastructure for Advanced Microbial Ecology Research and Analysis site (CAMERA) also maintains a repository of metagenomics sequence data at https://portal.camera.calit2.net/gridsphere/gridsphere, where we were able to download data from the Global Ocean Survey [#f1]_.
The human microbiome project has generated several terabytes of data from various locations on the bodies of a cohort of health people, which is described in detail at http://http://www.hmpdacc.org/ and shotgun metagenomics sets can be downloaded at their ftp site by, for example, typing:
wget ftp://public-ftp.hmpdacc.org/Illumina/anterior_nares/*.tar.bz2
Since these data files are compressed, tarred files, it is necessary to untar and unzip the files (:command: tar -jxf SRS011105.tar.bz2) and merge the paired-end files (:command: cat SRS011105/*.fastq > SRS011105.fastq) and re-compress if desired (:command: gzip SRS011105.fastq) before analyzing with Sequedex.
5.2. Phylogenetic rollups¶
For users wanting to look at output data as rapidly as possible, we
supply three Excel spreadsheets with the collected results for
reference genomes chopped into 100 bp reads, the human microbiome data, environmental metagenomes. The reference genomes were obtained from the
completed and draft genomes at the NCBI ftp site, and are the same
genomes used in the Life2550 reference tree.
When comparing phylogenetic profiles of metagenomes, it is helpful to produce a matrix of phylogenetic profiles, with the 2550 rows corresponding to the nodes on the bacterial phylogeny, and each sample assigned its own column, as well as a matrix of functional profiles, with the 963 rows corresponding to the SEED (http://www.theseed.org) functional categories. This can be done in many ways, including reading each output file into Excel and pasting the relevant columns into one sheet, but a simple shell-script will also do the job. If the user is comparing more than a handful of files, he or she will likely want to write a program to both combine the data files together and associate simplified labels with the longer, unique, labels assigned to ensure proper sample tracking. A simple script that does this is:
cd hmb_data/output
for i in SRR059330 SRR059331 SRR059338 SRR059339; do
awk '{print $2}' < $i.fq.sqdx/who-Life2550-16GB.tsv > $i.j;
awk '{print $2}' < $i.fq.sqdx/what-Life2550-16GBxseed_0911.m1.tsv > $i.k;
done
paste SRR059330.j SRR059331.j SRR059338.j SRR059339.j > ../hmb.stool.who
paste SRR059330.k SRR059331.k SRR059338.k SRR059339.k > ../hmb.stool.what
echo "stool1 stool2 stool3 stool4" > ../stool.lbl
This will combine the output files from four stool metagenomics data sets into a matrix of counts for the who and what files. The user will, of course, need to supply directory and file names corresponding to his particular analysis. A spreadsheet may help to track the relationships between the sample names and labels. Note also that the paste command by default places tab characters between columns, while the echo command in the above script is placing spaces between labels. For the 100 bp synthetic data made from each of the 2405 bacterial and archeal genomes, with the results sorted phylogenetically and rolled up to the phylum level, one obtains:
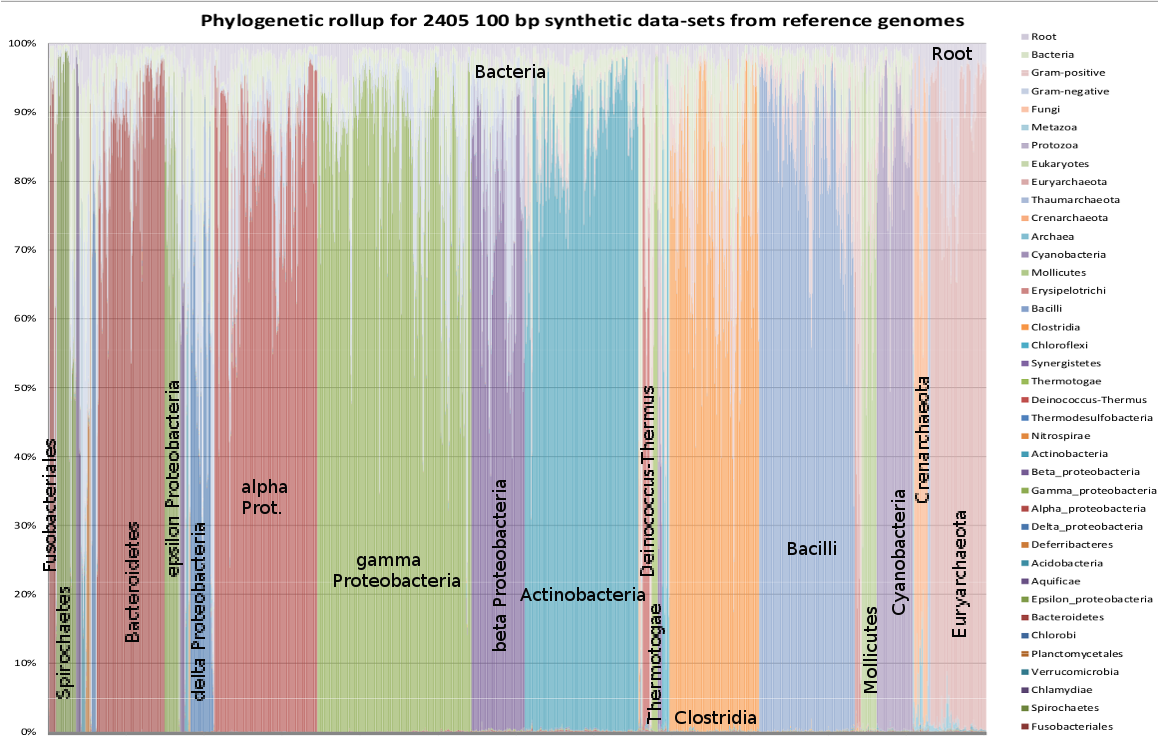
When combined with the phylogenetic and data module size dependence of the fraction of reads recoginzed, shown in Alternative data modules and memory usage, the user can see that well-studied portions of the phylogeny, typically > 80 % of the reads are classified, with > 90 % having reasonably specific assignment. The tax in these figures are in the same order as in Tree of Life, 2550 taxa. The tools to explore phylogenetic assignments will be explored in much greater detail in the next chapters.
From these raw counts files, several types of profiles can easily be
computed: phylogenetic rollups, normalized phylogenetic profiles, and
normalized scalar products of the profiles. Once again, these
quantities can be computed in many ways, but the gfortran
(http:/gcc.gnu.org/wiki/GFortran/) code will suffice, and is described at the end of
The command line with standard tools (Linux, Mac, Cygwin for Windows). In particular, examination of the tree with archyoptrix (http://www.phylosoft.org) or
an output file
reveals forty roll-up categories, defined in the notation of Fortran
90 here. Only eight of the most
highly represented categories are shown in the human microbiome plot,
while the environmental metagenomes rollup, below, shows seventeen
categories, with only ‘multiphyla’ and ‘root’ left off.
A description of the project aims and primary analysis results can be found in “A framework for human microbiome research” by the Human Microbiome Consortium, in http://www.nature.com/nature/journal/v486/n7402/full/nature11209.html.
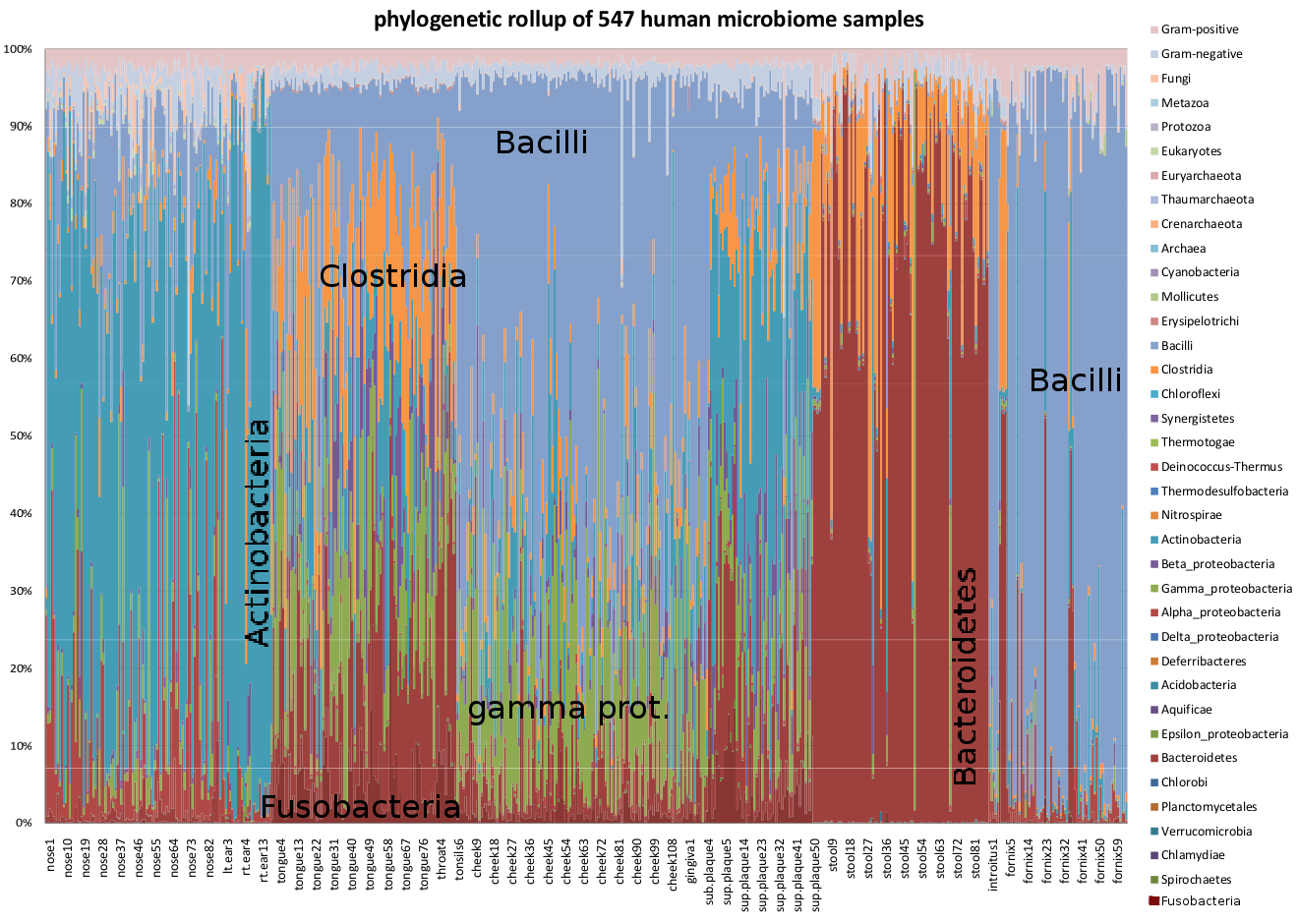
Phylogenetic profile of 547 human microbiome samples, computed
using the Life2550-40GB data module and Sequedex, rolled up with
the above-defined classifications. The original plot and data can
be found in the first panel of this Excel spreadsheet. For the most parts, the samples consist of
pairs of replicates, and they are nearly indistinguishable from
each other in this presentation.
The phylogenetic and functional profiles presented here can be compared to Figure 2 of “Structure, function, and diversity of the health human microbiome”, also by the Human Microbiome Consortium, in http://www.nature.com/nature/journal/v486/n7402/full/nature11234.html. Although the Human Microbiome Consortium ran their datasets through BLASTX (see SRS016585 at http://www.hmpdacc.org/HMSCP/#data to verify that stool sample run SRR059346 is indeed primarily E. coli), the analysis in the HMB paper utilized a rapid-matching scheme, described in Segata, et al., “Metagenomic microbial community profiling using unique clade-specific marker genes” Nature Methods 9:811-814 (2012) http://www.nature.com/nmeth/journal/v9/n8/full/nmeth.2066.html.
Although Segata, et al. used clade-specific genes to profile metagenomics data, while we used phylogenetic signatures of the majority of genes, numerous points of agreement between the analyses are evident. Note that we included a ‘multiphyla’ category in our figure, which was not included in their reported results. Specifically:
- For the stool samples, the ratio of bacteroidetes to clostridia varies from 60% clostridia to 95% bacteroidetes.
- For the tongue dosrum samples, firmicutes range from half to 10% of the identified species, with proteobacteria next most abundant, followed by bacteroidetes.
- For the buccal mucosa samples, firmicutes are even more prevalent, together with proteobacteria making up more than 90% of the assigned reads.
- For the supragingival plaque samples, roughly equal representation of firmicutes, actinobacteria, proteobacteria, and bacteroidetes occurs, with considerable variability in the relative abundance.
- For the anterior nares samples, actinobacteria makes up more than 90% of the assigned reads in some samples, with the firmicutes providing the bulk of the balance.
- For the posterior fornix samples, most of the samples consist of firmicutes (clostridia + bacilli), although three of the analyzed by Sequedex consisted of equal parts bacteroidetes and actinobacteria, as did one in Figure 2 of the HMB paper.
- For the retroauricular crease samples, most of the samples are almost entirely actinobacteria, while alpha proteobacteria make up a significant minority of several of the samples.
We will return to further analysis of the HMB dataset after examining a set of representative environmental microbiomes.
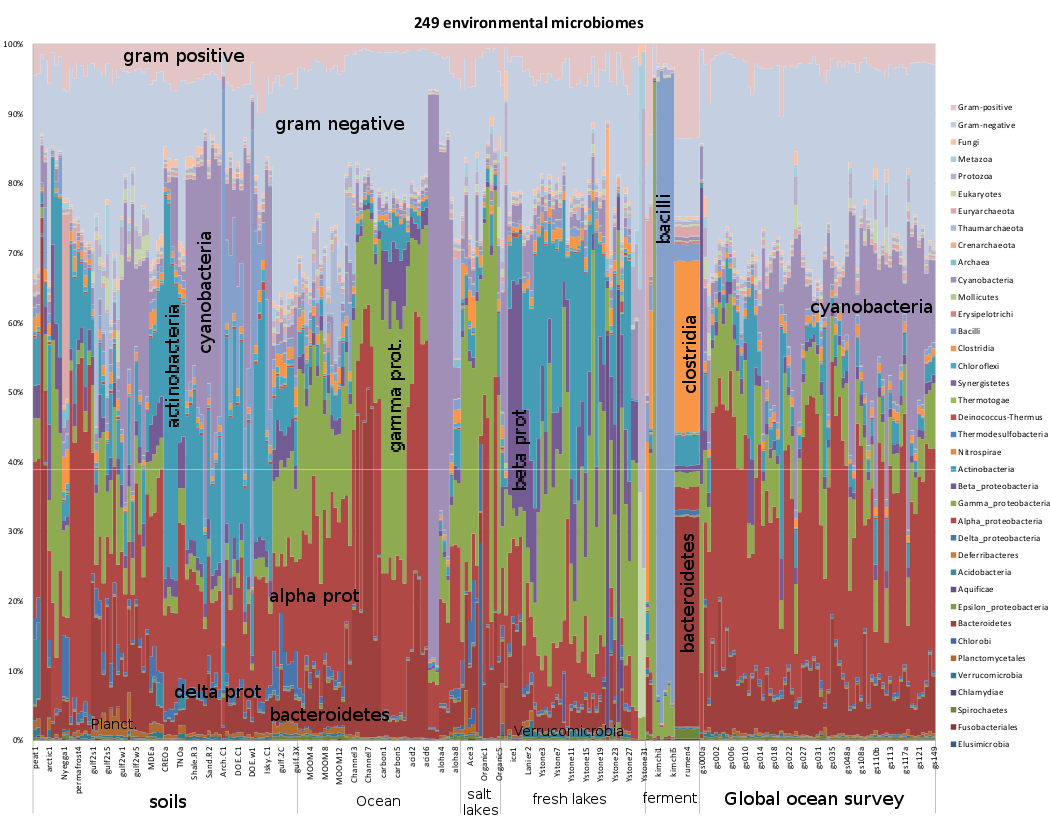
Phylogenetic profile of 249 environmental microbiome samples,
computed using the Life2550-40GB data module and Sequedex, rolled
up with the above-defined classifications. The original plot and
data can be found in the first panel of this Excel
spreadsheet. The data are a compilation of 27
distinct studies, with citations provided below, in order from left
to right on the figure. The data come primarily from the sequence
read archive (indicated by the SRR number above the relevent
columns in the spreadsheet. The Global Ocean Survey results can be
obtained from CAMERA (https://portal.camera.calit2.net/) and the
FACE data from JGI/IMG (http://img.jgi.doe.gov/), as well as
several data sets kindly provided before publication and release by
Cheryl Kuske at Los
Alamos National Laboratory.
A cursory examination of the environmental microbiome profiles reveals distinctive and repeatable differences between studies. We present here references to the publications from each sample, and a brief summary of salient details.
peat Kanokratana, et al., “Insights into the phylogeny and metabolic potential of a primary tropical peat swamp forest microbial community by metagenomic analysis” Microb. Ecol. 61:518-528 (2011). (http://www.ncbi.nlm.nih.gov/pubmed/21057783)
Permafrost1 Yergeau E, Hogues H, Whyte LG, Greer CW. “The functional potential of high Arctic permafrost revealed by metagenomic sequencing, qPCR and microarray analyses.” ISME J. 2010 Sep;4(9):1206-14. (http://www.ncbi.nlm.nih.gov/pubmed/20393573)
arctic Yergeau E, Sanschagrin S, Beaumier D, Greer CW, “Metagenomic Analysis of the Bioremediation of Diesel-Contaminated Canadian High Arctic Soils. PLoS ONE 7:e30058 (2012) (http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0030058)
Nyegga Stokke R, Roalkvam I, Lanzen A, Haflidason H, Steen IH., “Integrated metagenomic and metaproteomic analyses of an ANME-1-dominated community in marine cold seep sediments” Environ Microbiol. 2012 May;14(5):1333-1346 (2012) (http://www.ncbi.nlm.nih.gov/pubmed/22404914). Anaerobic methanotrophic archaea (ANME) and sulfur metabolising bacteria presumably include the delta proteobacteria visible in this sample.
Harvard FJ Stewart, AK Sharma, JA Bryant, JM Eppley, and EF DeLong “Community transcriptomics reveals universal patterns of protein sequence conservation in natural microbial communities” Genome Biology 12:R26 (2011). (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3129676/)
permafrost2 Mackelprang R, Waldrop MP, DeAngelis KM, David MM, Chavarria KL, Blazewicz SJ, Rubin EM, Jansson JK. “Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw.” Nature. 2011 Nov 6;480(7377):368-71. (http://www.ncbi.nlm.nih.gov/pubmed/22056985)
gulf2 Widger, et al., “Longitudinal Metagenomic Analysis of the Water and Soil from Gulf of Mexico Beaches Affected by the Deep Water Horizon Oil Spill” Preceedings of Nature hdl:10101/npre.2011.5733.1 (http://precedings.nature.com/documents/5733/version/1). The first eight samples are from sand, the second eight are from water.
face Berendzen, et al, “Rapid phylogenetic and functional classification of short genomic fragments with signature peptides” BMC Research Notes 5:460 (2012). (http://www.biomedcentral.com/1756-0500/5/460/abstract). For a description of the FACE sites, see http://public.ornl.gov/face/results.shtml.
utah Kuske, CR, et al., Soil crusts in Utah field site. manuscript in preparation.
MOOM Canfield, DE, et al., “A cryptic sulfur cycle in oxygen-minimum-zone waters off the Chilean coast” Science 330:1375 (2010).
E. channel Gilbert, JA, et al., “Metagenomes and metatranscriptomes from the L4 long-term coastal monitoring station in the Western English Channel” Standards in Genomic Sciences 3:183-193 (2010). Only functional analysis is presented, with SEED and MG-RAST.
carbon Mou, X., S. Sun, RA Edwards, RE Hodson, MA Moran “Bacterial carbon processing by generalist species in the coastal ocean” Nature 451:708-711 (2008). (http://www.nature.com/nature/journal/v451/n7179/abs/nature06513.html)
acid Gilbert JA, Thomas S, Cooley NA, Kulakova A, Field D, Booth T, McGrath JW, Quinn JP, Joint I., “Potential for phosphonoacetate utilization by marine bacteria in temperate coastal waters.” Environ Microbiol. 11:111-125 (2009) (http://www.ncbi.nlm.nih.gov/pubmed/18783384)
aloha Martinez A, Tyson GW, Delong EF., “Widespread known and novel phosphonate utilization pathways in marine bacteria revealed by functional screening and metagenomic analyses.” Environ Microbiol. 12:222-238 (2010) (http://www.ncbi.nlm.nih.gov/pubmed/19788654)
Ace Lake Lauro, FM, et al., “An integrative study of a meromictic lake exosystem in Antarctica” ISME J. 5:879-895 (2011) (http://www.nature.com/ismej/journal/v5/n5/abs/ismej2010185a.html)
Organic Lake Yau, S., et al. “Virophage control of antarctic algal host–virus dynamics” PNAS, USA 108:6163-6168 (2011). (http://www.pnas.org/content/early/2011/03/24/1018221108.full.pdf+html) Primarily about viruses.
stromatolites Desnues C., et al., “Biodiversity and biogeography of phages in modern stromatolites and thrombolites” Nature 452:340-345 (2008) (http://www.bio.sdsu.edu/faculty/kelley/31.pdf)
fanning Dinsdale, EA., et al., “Microbial ecology of four coral atolls in the Northern Line Islands” PLoS One. 3:e1584 (2008). (http://www.plosone.org/article/info:doi/10.1371/journal.pone.0001584)
wastewater Sanapareddy, N., et al., “Molecular Diversity of a North Carolina Wastewater Treatment Plant as Revealed by Pyrosequencing” Appl Environ Microbiol. 75:1688–1696. (2009). (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2655459/)
ice Simon, C., A. Wiezer, AW Strittmatter, R. Daniel, “Phylogenetic Diversity and Metabolic Potential Revealed in a Glacier Ice Metagenome” App. & Env. Microb. 75:7519-7526 (2009). (http://aem.asm.org/content/75/23/7519.full). In agreement that the major phyla are Betaproteobacteria, Bacteroidetes, and Actinobacteria.
Lake Lanier Oh, S., et al., “Metagenomic insights into the evolution, function and complexity of the planktonic microbial community of Lake Lanier, a temperate freshwater ecosystem” App. & Env. Microbiol. doi: 10.1128 (2011). (http://aem.asm.org/content/early/2011/07/15/AEM.00107-11.short).
Yellowstone Inskeep, WP., “Metagenomes from High-Temperature Chemotrophic Systems Reveal Geochemical Controls on Microbial Community Structure and Function” PLoS ONE 5:e9773 (2010). (http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0009773). Not sure if this is the correct study.
biogas Jaenicke, S., et al. “Comparative and Joint Analysis of Two Metagenomic Datasets from a Biogas Fermenter Obtained by 454-Pyrosequencing” PLoS one 6:e4519 (2011) (http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0014519)
Leaf-cutter ants Aylward FO, et al. “Metagenomic and metaproteomic insights into bacterial communities in leaf-cutter ant fungus gardens” ISME J. 6:1688-701 (2012). (http://www.ncbi.nlm.nih.gov/pubmed/22378535)
kimchi Jung JY, Lee SH, Kim JM, Park MS, Bae JW, Hahn Y, Madsen EL, Jeon CO, “Metagenomic analysis of kimchi, a traditional Korean fermented food” Appl Environ Microbiol. 77:2264-74 (2011). (http://www.ncbi.nlm.nih.gov/pubmed/21317261)
rumen Hess, M., et al., “Metagenomic discovery of biomass-degrading genes and genomes from cow rumen” Science. 331:463-7 (2011). (http://www.sciencemag.org/content/331/6016/463.short)
Global Ocean Survey Rusch, DB, et al. “The Sorcerer II global ocean sampling expedition: Northwest Atlantic through Eastern Tropical Pacific” PLoS Biology 5:e77(2007) (http://www.plosbiology.org/article/info:doi/10.1371/journal.pbio.0050077).
Another use for sequedex is to identify bacterial symbionts in eukaryotic samples, such as are readily visible in several of the transcripts from the Marine Microbial Eukaryote Transcriptome Sequencing Project.
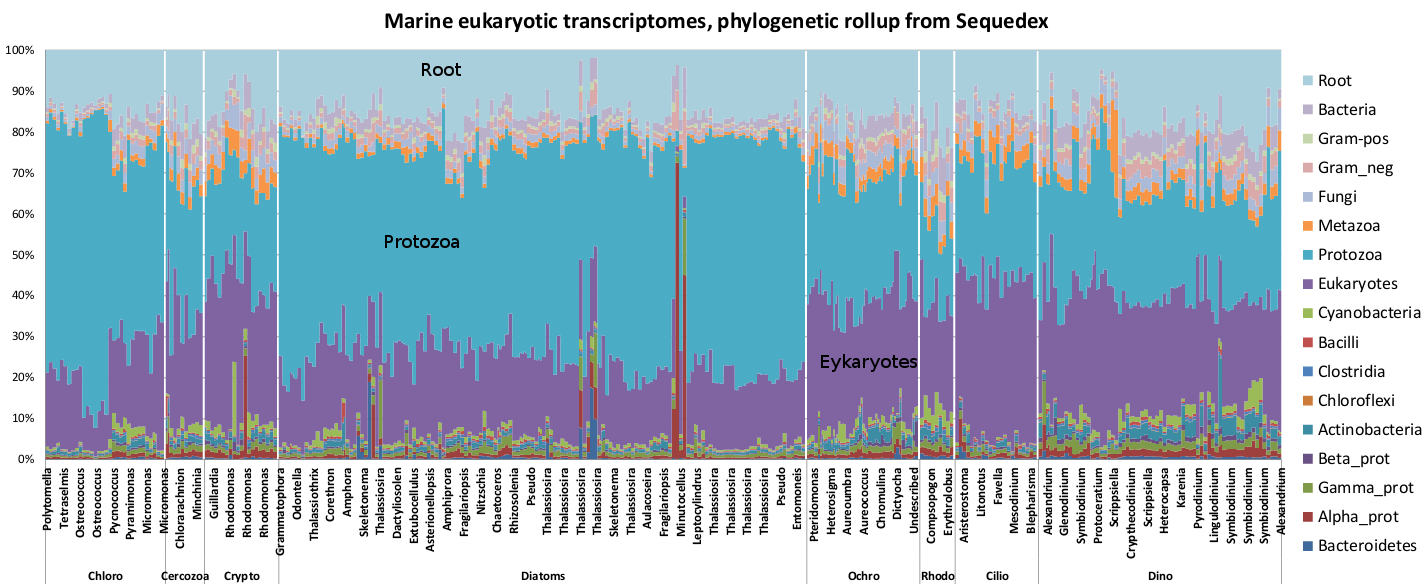
Phylogenetic rollup of microbial marine transcriptomes. Since most of the transcriptomes are from lower eukaryotes only distantly related to completed genomes, most of the reads are identified simply as ‘protozoal’ or ‘eukaryotic’. Nevertheless, the samples where reads are predominantly bacterial are easily visible in this figure.
5.3. Functional rollups¶
It is possible to produce functional profiles of the same samples used
above for comparing phylogenetic profiles. Since we are using the
SEED functional classification scheme, it is also possible to
‘roll-up’ the functional profiles into 28 categories of gene function.
Inspection of the output file SRR059330.fasta.fun reveals the
following roll-up categories, defined in the notation of Fortran 90
here.
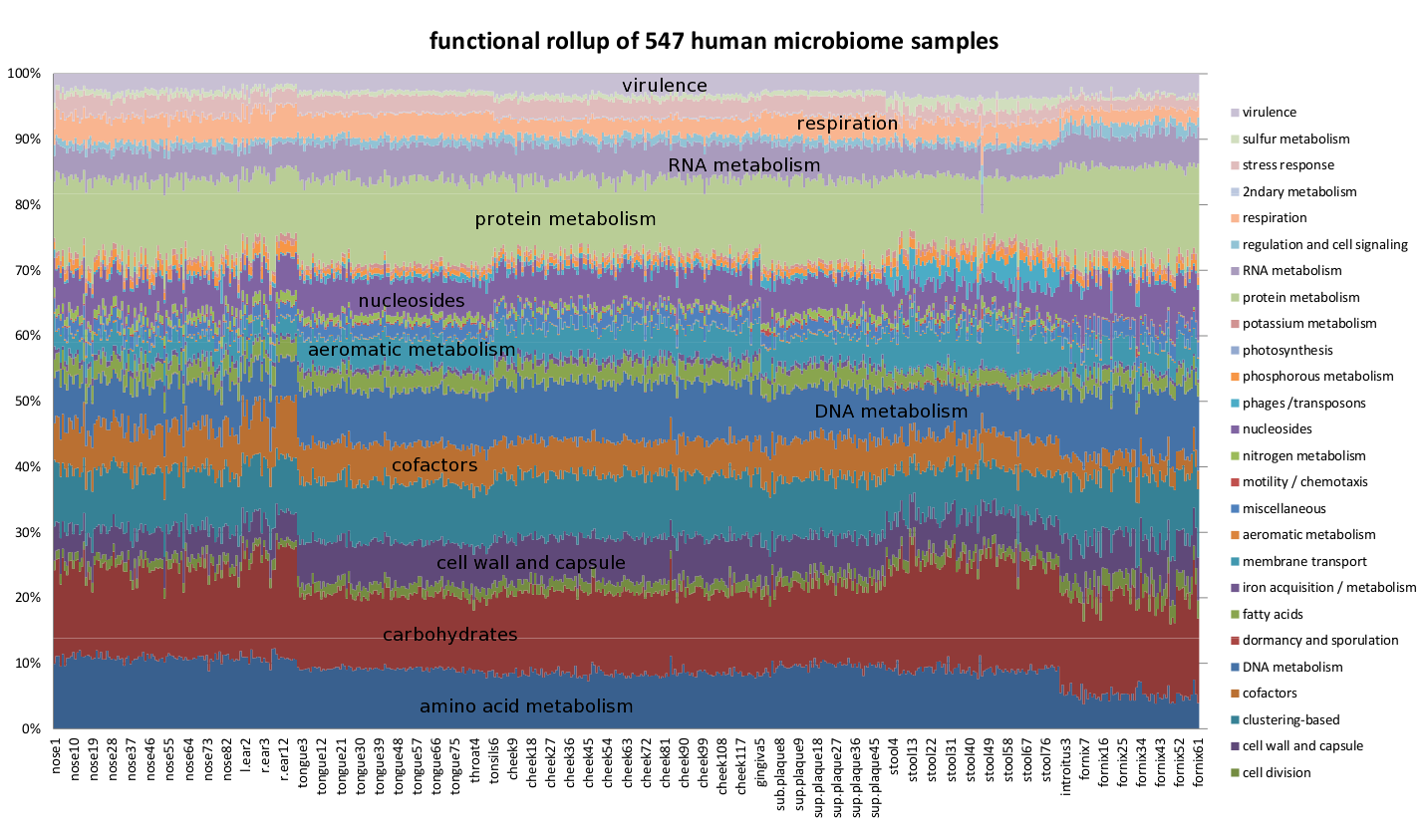
Functional profile of 547 human microbiome samples, computed using the Life2550 tree, the SEED classification, and Sequedex, rolled up with the above-defined classifications. The functional profiles are provided above in an Excel spreadsheet.
The most striking observation about the functional profile across the 547 human microbiome samples is the consistent relative amplitude of the different functions from the different body sites. The two sample types with the greatest variation in functional rollup profiles are the posterior fornix and the anterior nares, consistent with Figure 2 of the Nature article referenced above (Nature 486:207-214 (2012)). It is possible that this variability is a consequence of clonal populations of particular organisms that make up a significant population of some of the samples skewing the results, but we have not explored this idea further. We observe in the environmental functional profiles, below, that these relative amplitudes are also broadly in agreement across diverse ecosystems. We will return to this discussion below, when we examine the functional profiles in greater resolution.
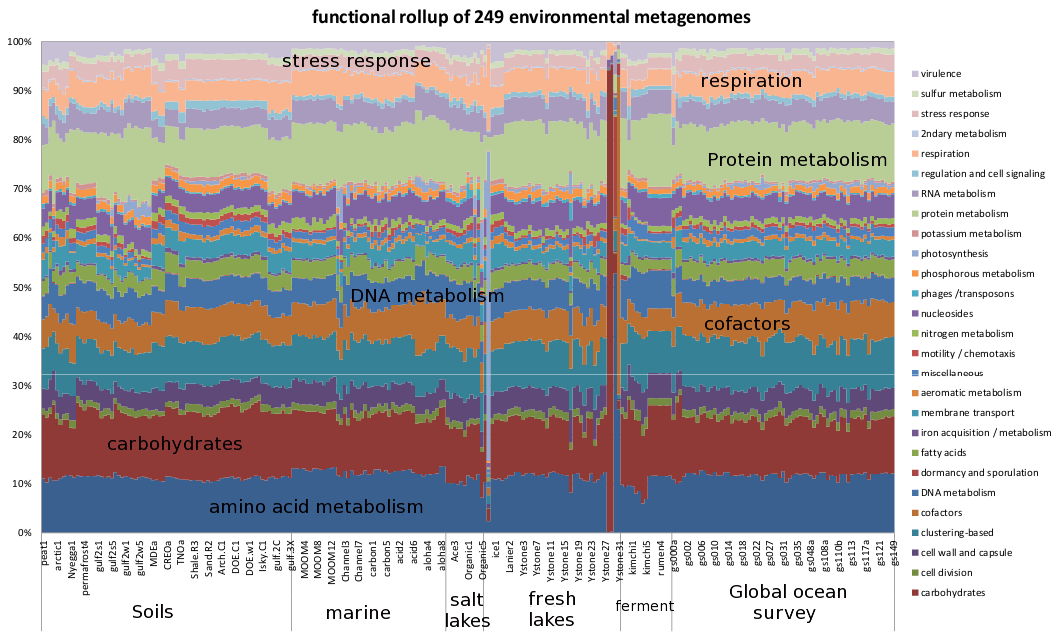
Functional profile of 249 environmental microbiome samples with the same color-scheme as the HMB functional profile above, computed using the Life2550 tree, the SEED classification, and Sequedex, rolled up with the above-defined classifications. The functional profiles are provided above in an Excel spreadsheet. As with the human microbiome samples, the consistent relative fraction of each functional rollup is quite striking, as is the enormous dispersions that arise from this uniformity.
The twenty functional categories with more than 1000 counts in the TNO sample are shown below with the subsystem number from the figure and the three levels of annotation. The top two levels are shortened to help make the lowest level fit on the page. Genes in each SEED subsystem are found by clicking on the subsytem title in section Definition of functional classifications. Many of the subsytems also have detailed explanations available at http://www.theseed.org.
si_0032 Amino Acids Glutamine, GLN, ASP, ASN; ammonia Glutamine,_Glutamate,_Aspartate_and_Asparagine_Biosynthesis
si_0042 Amino Acids Lysine, threonine, MET, Cys Methionine_Biosynthesis
si_0078 Carbohydrates Central carbohydrate metabolism Pyruvate_metabolism_II:_acetyl-CoA,_acetogenesis_from_pyruvate
si_0122 Carbohydrates One-carbon Metabolism Serine-glyoxylate_cycle
si_0205 Cell Wall Unclassified cell wall and capsule Peptidoglycan_Biosynthesis
si_0279 Clustering Unclassified clustering-based Bacterial_Cell_Division
si_0334 Clustering Unclassified clustering-based Conserved_gene_cluster_associated_with_Met-tRNA_formyltransferase
si_0357 Cofactors Biotin Biotin_biosynthesis
si_0415 DNA Meta. DNA replication DNA-replication
si_0448 Fatty Acids Fatty acids Fatty_Acid_Biosynthesis_FASII
si_0526 Membrane tra. Unclassified membrane transport Ton_and_Tol_transport_systems
si_0589 Nitrogen Unclassified nitrogen metabolism Ammonia_assimilation
si_0602 Nucleosides Purines Purine_conversions
si_0640 Phosphorus Unclassified phosphorus metabolism Phosphate_metabolism
si_0654 Potassium Unclassified potassium metabolism Potassium_homeostasis
si_0660 Protein Protein biosynthesis Ribosome_LSU_bacterial
si_0790 Regulation Unclassified regulation and cell sig cAMP_signaling_in_bacteria
si_0812 Respiration Electron donating reactions Respiratory_Complex_I
si_0929 Virulence Resistance to antibiotics and toxic Cobalt-zinc-cadmium_resistance
si_0939 Virulence Resistance to antibiotics and toxic Multidrug_Resistance_Efflux_Pumps
Functional rollups from genomic (DNA) transcriptomes have similar quantities of reads from each of the functional rollup categories. Examination of the functional rollup of the marine algae transcriptomes, however, show much more variable, and results that depend on growth conditions.
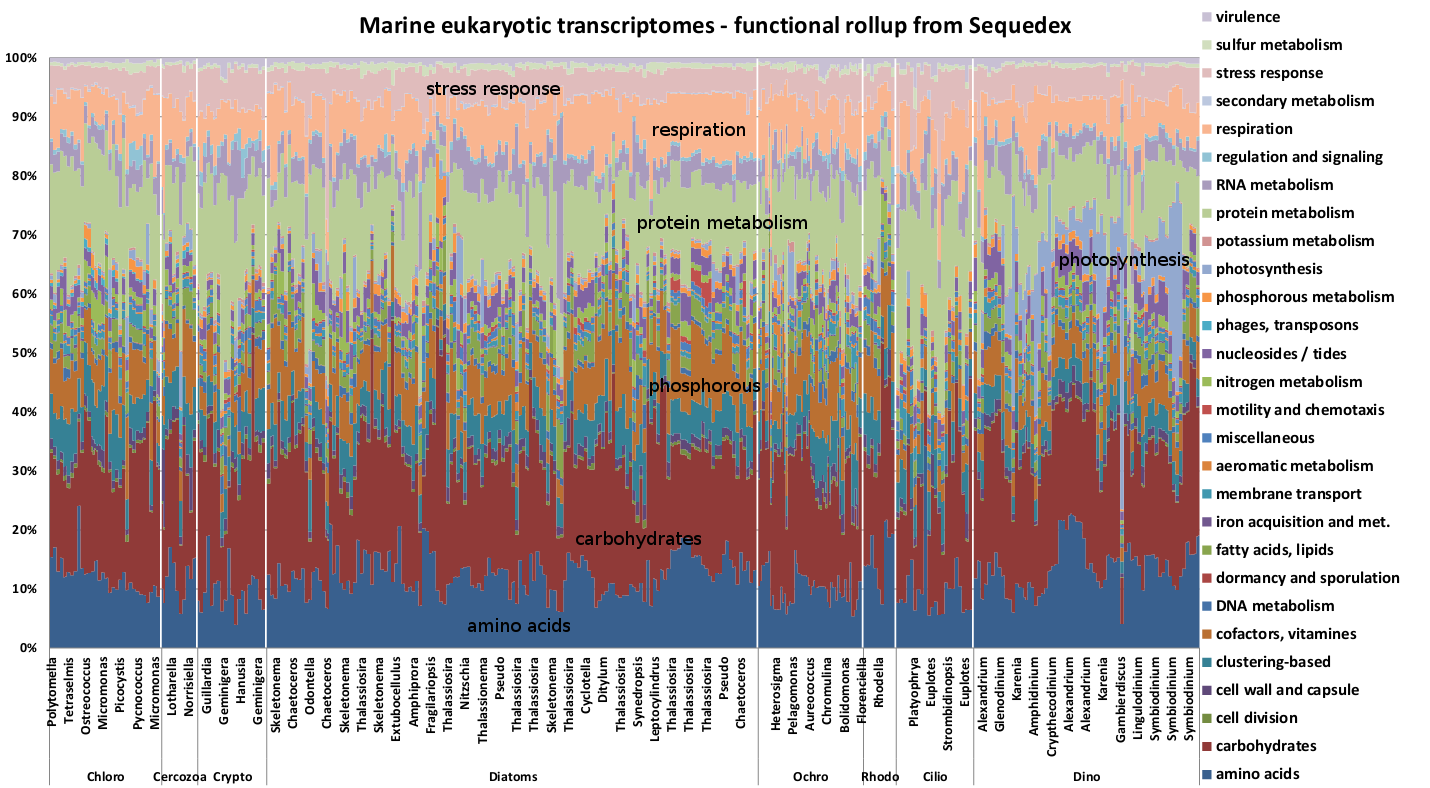
Functional rollup of 330 marine eukaryotic algal transcriptomes. The transcriptomes are grouped phylogenetically, on the basis of the small subunit ribosomal RNA sequence obtained from the sample. It is evident that the condition-dependence of the functional rollups are significant, but the correlations of conditions with functional rollup will require closer examination.
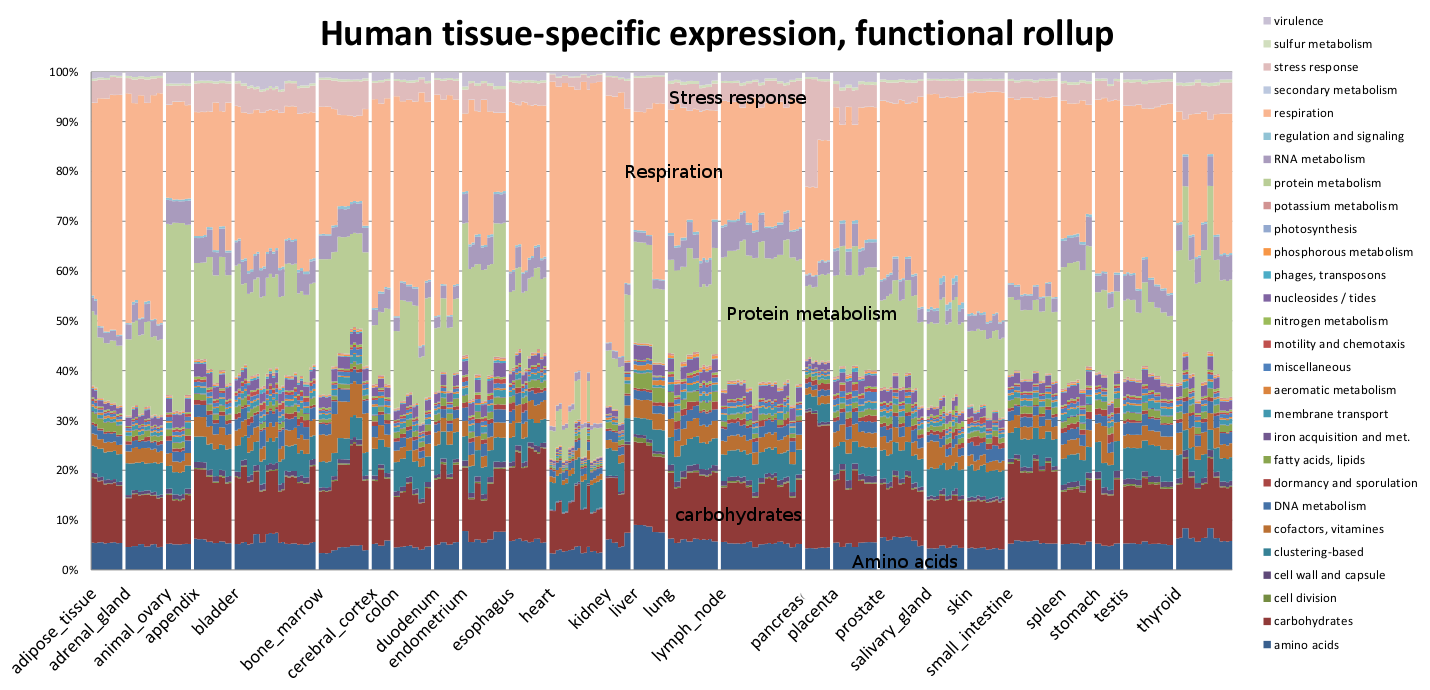
When the transcriptomes are taken from a single organism and grouped by condition, such as with the Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics, by Fagerberg, *et al.*, the statistically significant differences in functional expression by grouping are immediately evident.
5.4. Normalized functional profiles¶
To investigate further, we examine the functional profiles of the larger data sets for the 400 human microbiome samples and 242 enviromental samples for individual SEED subsystems, answering the question, ‘What type of nitrogen metabolism is present?’ rather than ‘Does the organism metabolise nitrogen?’.
Since most of the SEED subsystems cover metabolic and other functions necessary for all organisms, the consistency across samples is more reassuring than informative. To convince ourselves that the functional profiles are capable of distinguishing samples from one-another, we normalize the functional counts (not including ‘unassigned function’ and compare two categories that are observed to vary across environmental and human microbiome samples.
Normalization occured with Fortran 90 code; see F90 code. One of the easiest functional categories to
interpret is photosynthesis across the environmental metagenomes. By
comparison, none of the human microbiome samples contained more than
0.25% photosynthesis, and this was primarily composed of hits to
proteorhodopsin, rather than the photosystems.
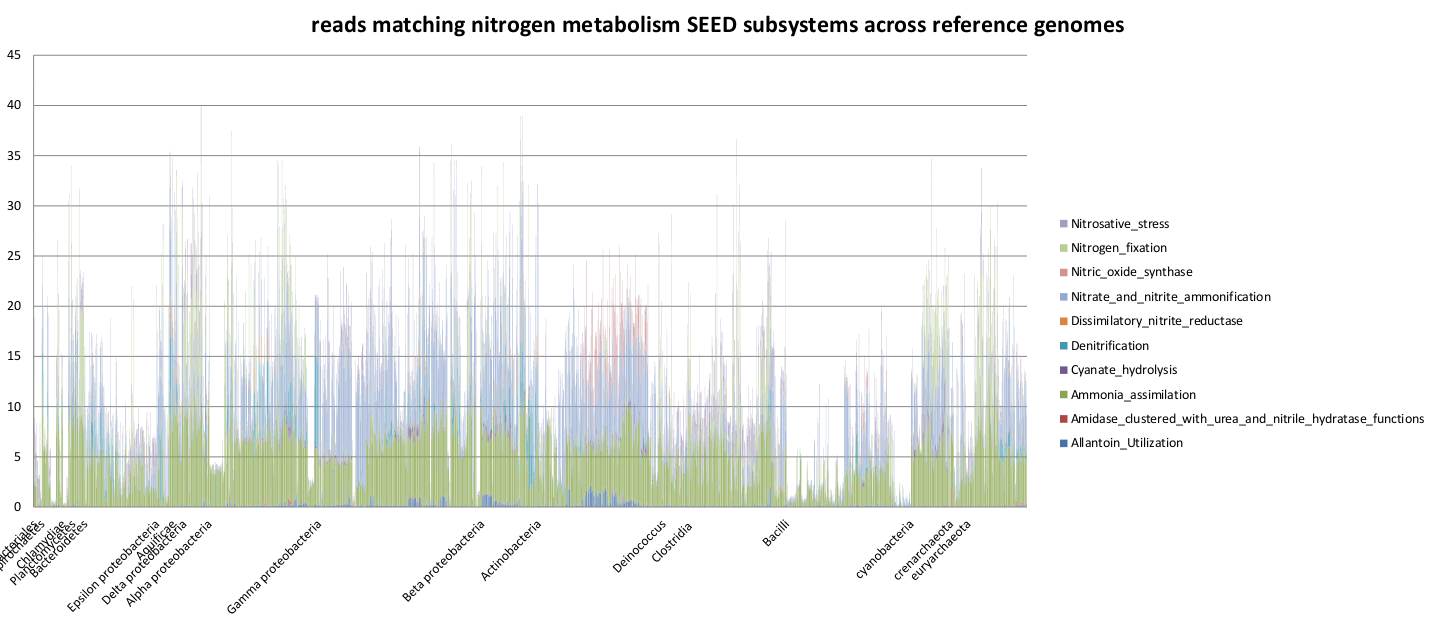
Fraction of the functionally assigned reads encoding for nitrogen subsystems across the 2415 synthetic reference metagenomes.
When this figure is expanded and combined with insight from the microbiolgist, numerous useful insights can be gained, such as in these expanded views of three sets of subsystems across the cyanobacterial phylum, as in Proteomic profiles of five strains of oxygenic photosynthetic cyanobacteria of hte genus *Cyanothece*, by Aryal, *et al.*.
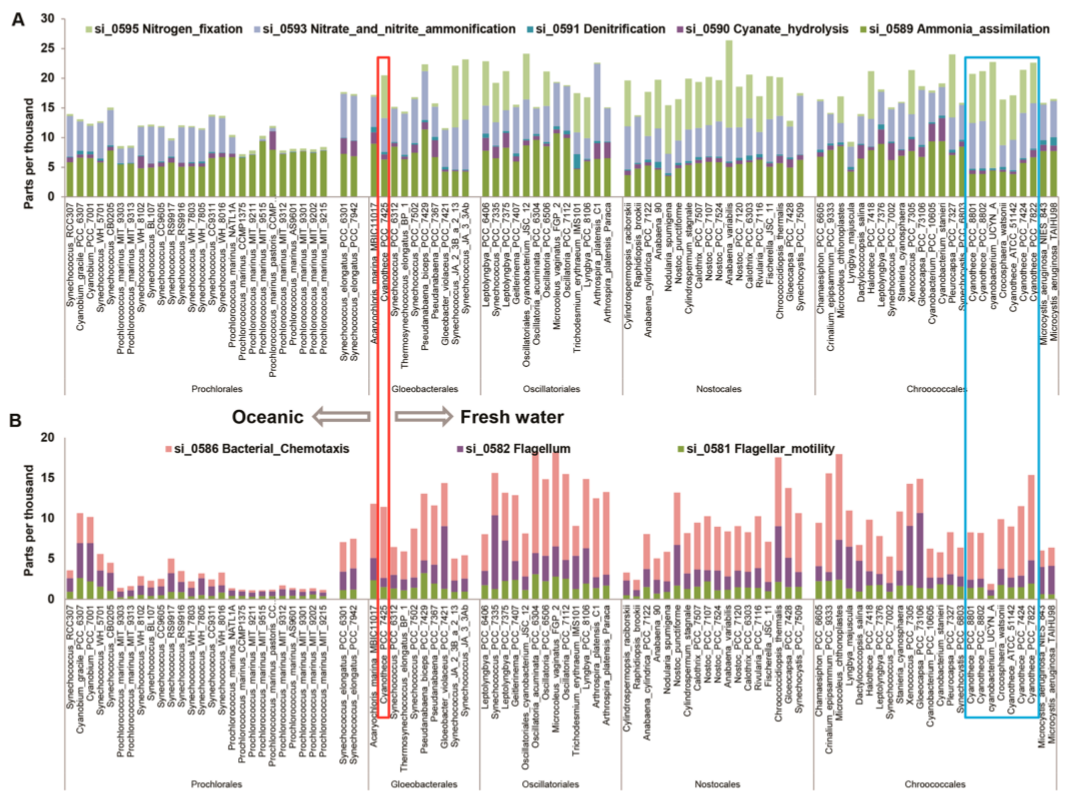
Distribution of genes involved in various cellular functions across 94 cyanobacterial genomes. Shown are the genes involved in nitrogen metabolism (A) and motility and chemotaxis (B). The data were normalized within each genome so that the sum of all functional categories adds to 1000 counts, making the units on the amplitudes of each function parts per thousand. Genes associated with the Cyanothece are shown in boxes.
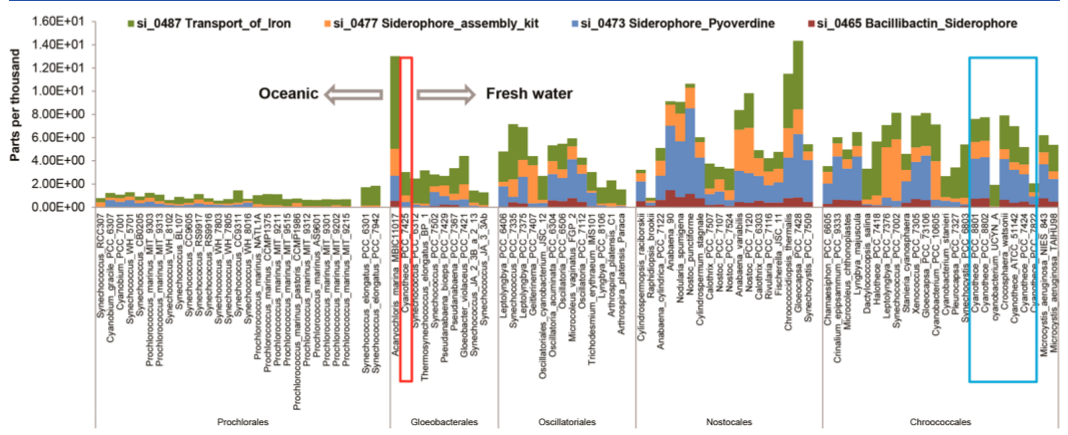
Distribution of genes involved in iron acquisition and transport across 94 cyanobacterial genomes. Genes associated with different Cyanothece species are shown in boxes.
For the environmental metagenomes, we chose to examine the photosynthesis.
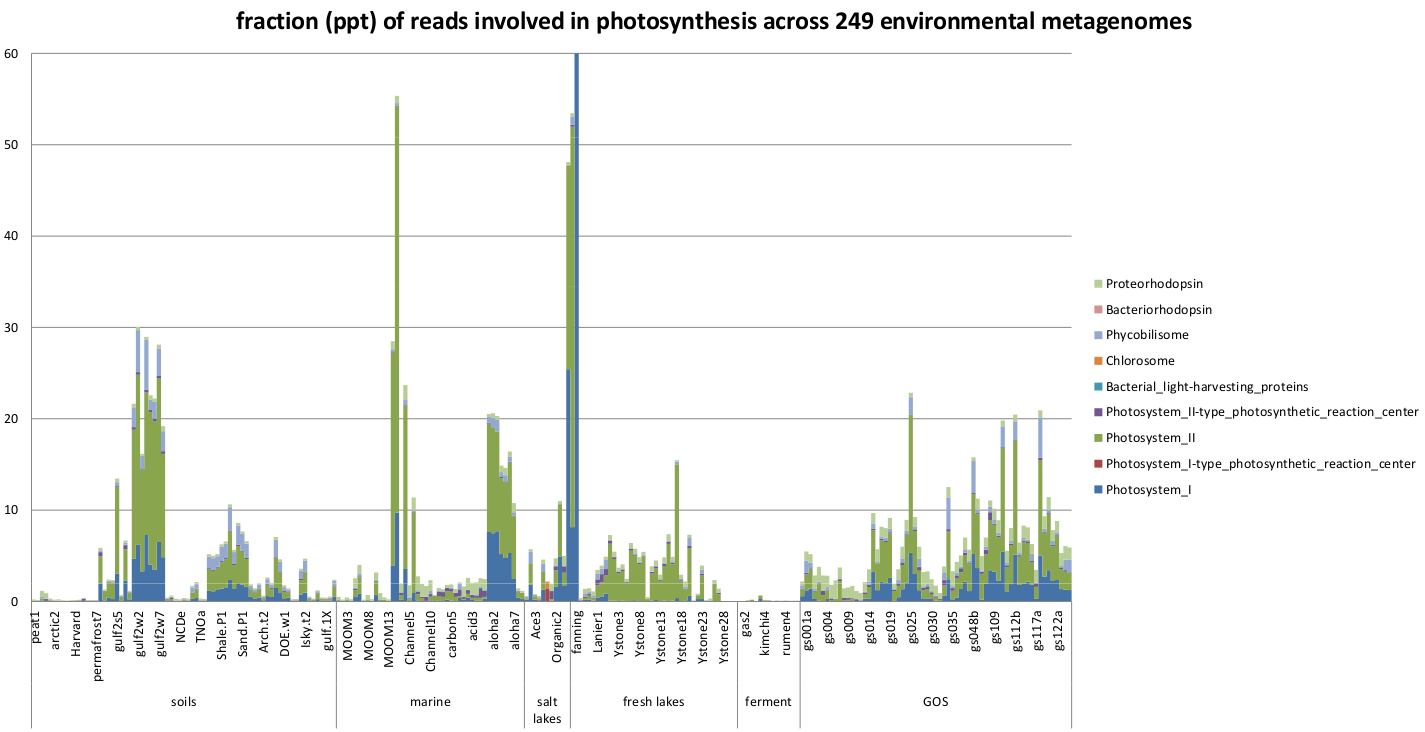
Fraction of the functionally assigned reads encoding for phytosynthesis subsystems across the 249 environmental metagenomes.
For the human microbiome samples, we chose to examine the motility genes.
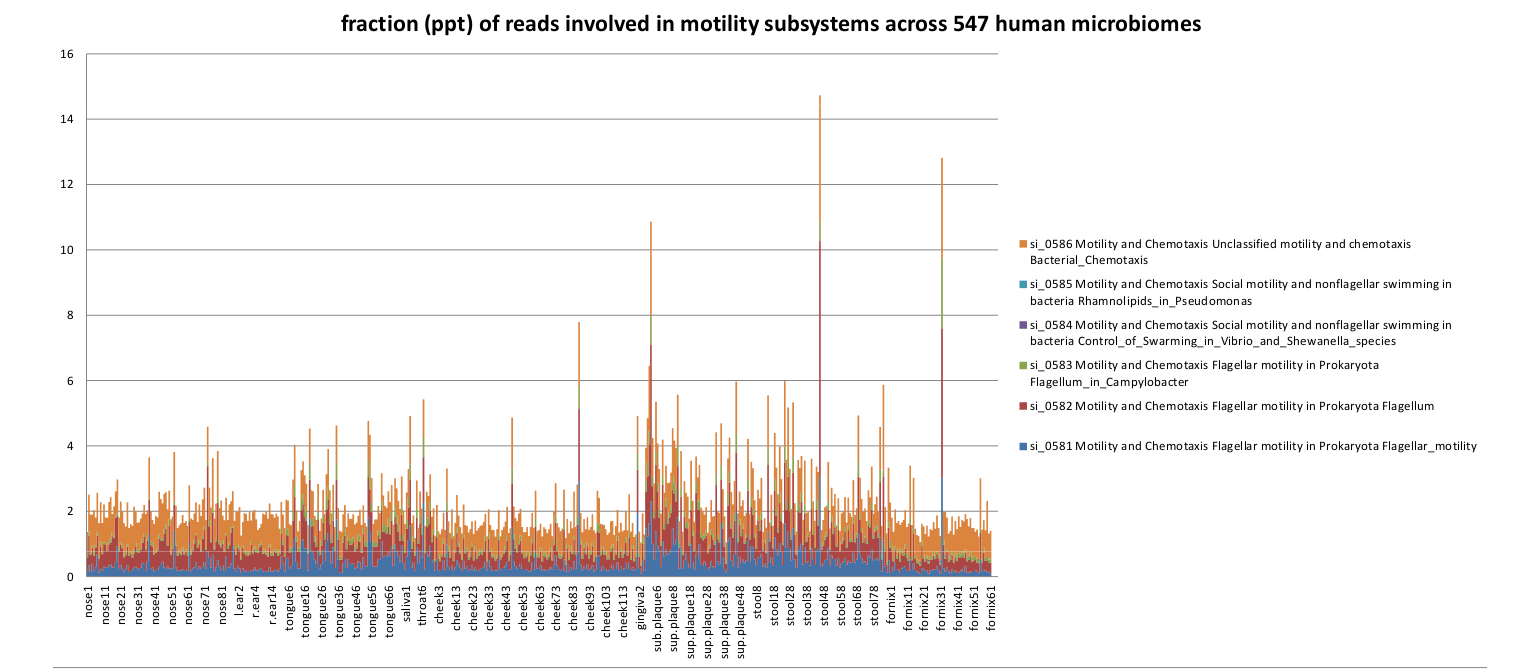
Fraction of the functionally assigned reads encoding for phytosynthesis subsystems across the 527 human microbiome metagenomes.
The set of output files can be viewed in Excel, and we supply two
Excel spreadsheets with the collected results for the human microbiome
data hmb.xlsx and environmental
metagenomes env.Life.xlsx. The tab
‘fnorm’ in each spreadsheet contains the normalized functional counts.
It is straightforward to scroll through this spreadsheet graphing
various subsystems of interest, or sorting the functional categories
based on counts in particular samples or difference in counts across
various samples. With some thought, it is also possible to sort based
on the signal-to-noise ratio with which the functional profiles
distinguish ecosystems, with the noise defined by replicate samples.
Examination of the SEED subsystems from the human tissue transcriptomes shows the tissue-specific espression in more detail.
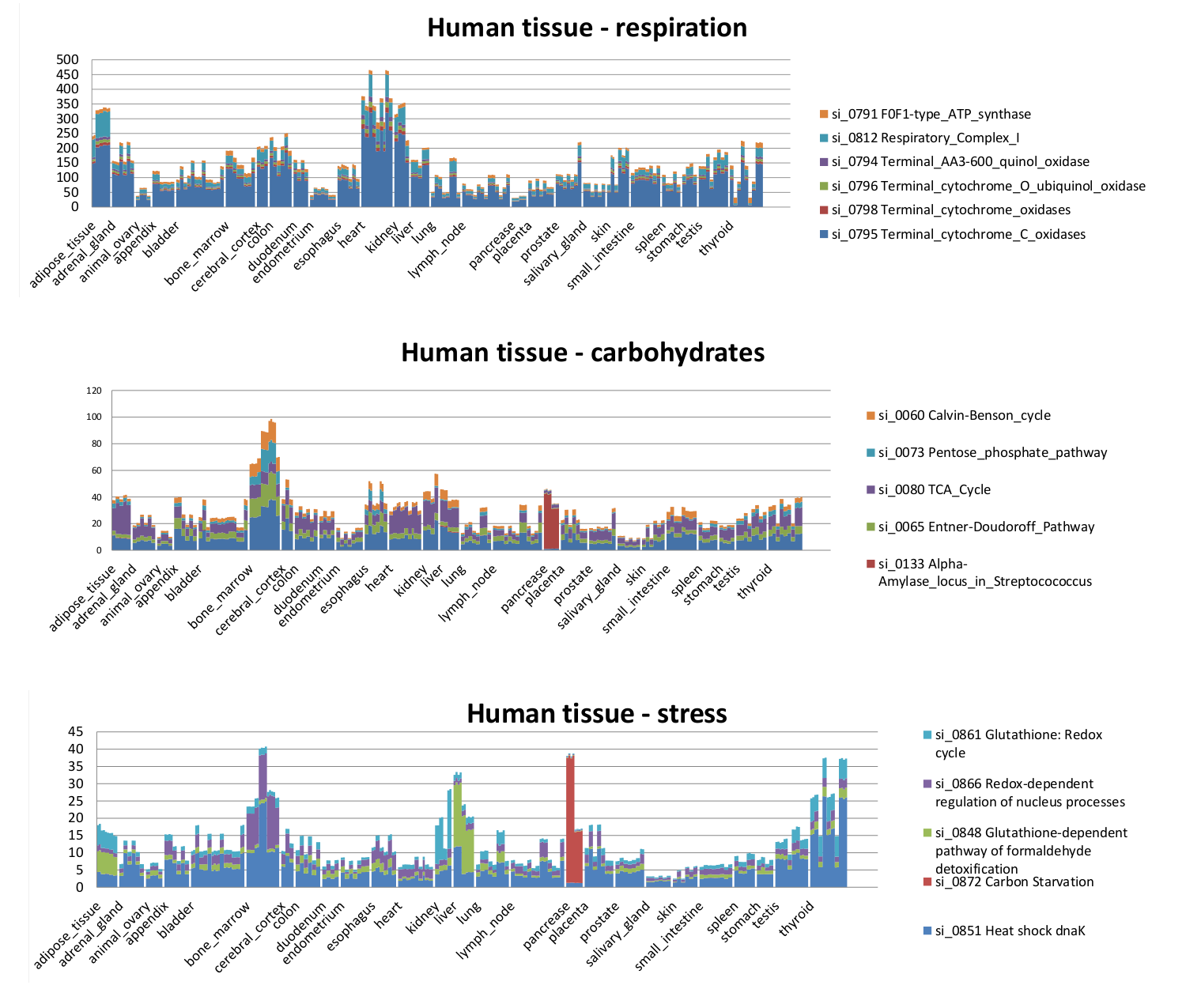
Normalized counts for the tissue-specific expression.
Thus, normalized functional profiles across reference data sets enable a ‘top-down’ approach to understanding functional classifications. The user may want to import results from their own metagenomics data sets into these spreadsheets to better understand the significance of their own results.