15. Reference data¶
As explained in the How is Sequedex used with other software? and described in some detail in Annotated reads, further analysis of closely related sequences can occur with reference alignments and tree-building. Creation and annotation of reference sequence alignments is a laborious process that consumes a significant portion of effort at such specialized efforts as the HIV sequece database or the Ribosomal Database project. We are not attempting to be comprehensive (for this, see Pfam or The Hemorrhagic Fever Viruses (HFV) Database Project) or even particularly accurate.
Instead, we aim to enable the user to build off of our efforts at characterizing one important phylogenetic marker (RNA Polymerase) to illustrate the techniques and enable others to replicate and trouble-shoot our phylogenetic work in creating our reference tree. Additionally, we provide reference alignments for a variety of other problems, such as strain-ID of pathogens and distinguishing orthologs from paralogs in ascribing functional roles to proteins such as virulence factors, enzymes, or regulatory proteins.
Another advantage to the two-step process for prodcing phylogenetic or functional assignments is that the most recent information can be used in the latter step. Because Sequedex uses a database of orthogenomic signature peptides to identify reads for further analysis, improvements in the reference database (Sequedex data module) affect only the overall sensitivity, and not the resolution of placement. Depending on the question being asked, the sensitivity may already be quite high (> 90%).
These alignments can be used in two ways to characterize an unknown; both require an alignment of metagenomic reads or assembled contigs to the reference alignment. For a relatively small number of aligned reads or contigs, the tree can simply be re-calculated, and the relative position of reference and sample can be compared. For a large number of aligned reads or contigs, p-values and Pplacer can be used to visualize which leaves or internal branches provide the most appropriate placement of the reads.
15.1. Phylogenetic placement of RNA Polymerase reads¶
RNAaPol alignment files, including beta and beta prime subunits for bacterial groups 1-5 of Tree of Life, 2550 taxa, beta, gamma, and beta prime subunits of the cyanobacteria, and the B1 and B2 subunits of eukaryotic RNA Pol II (care was taken to exclude RNA Pols I & III). The files are in Phylip format, because that is what is needed for Tree building with phyML or FastTree. It has the additional advantage that sub-alignments can easily be extracted by simply using the grep command:
grep Bacteroides data.1 > Bacteroides.RNAP.fas
will extract the 23 Bacteroides polymerase sequences into a separate file. This provides a convenient alignment to which assembled genes from metagenomes can be added, for the purpose of generating trees showing how organisms from the metagenomic samples relate both to the reference species and one another.
Sequence file formats can be converted online at the HIV database.
Subsets of the above files generated the bacterial reference trees used by Sequedex. The above seven groups cover the bacteria and archaea - for the eukaryotes, RNAPol II must be separated from RNAPols I and III (data not provided here) to create a tree based on RNAPol II, such as this:
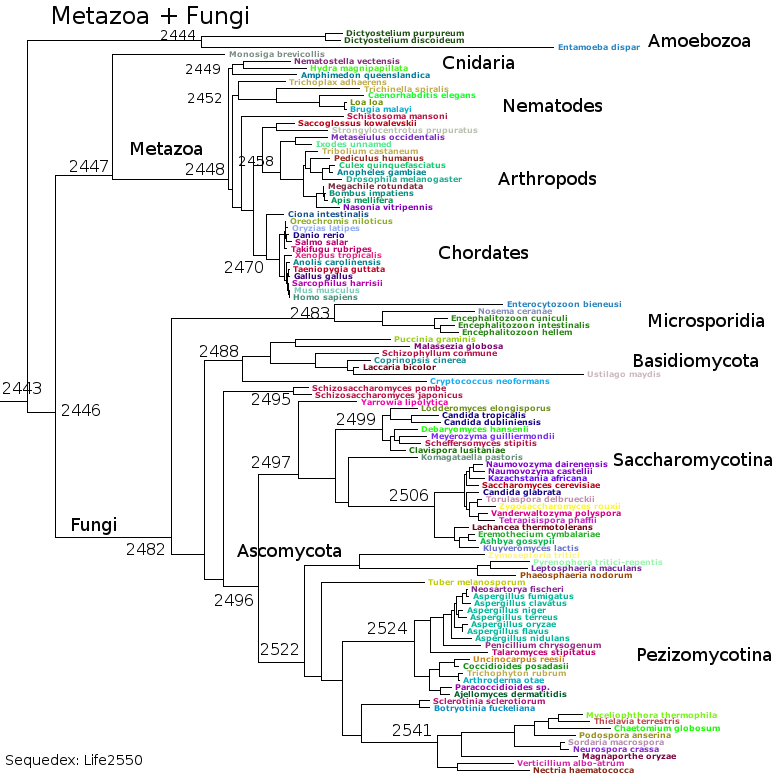
Maximum likelihood tree of the fungi and metazoa computed with PhyML from an amino acid alignment of the B1 and B2 genes of RNA Polymerase II.
15.2. Nucleotide Alignments for Strain Attribution¶
By using nucleotide alignments and all available strains from NCBI’s completed and draft genomes instead of amino acid alignments and one representative per species, it is possible to see considerable structure in the various strains of different species, as well as interspersing of strains from two different species. The figure below was made from near-neighbors of E. coli.
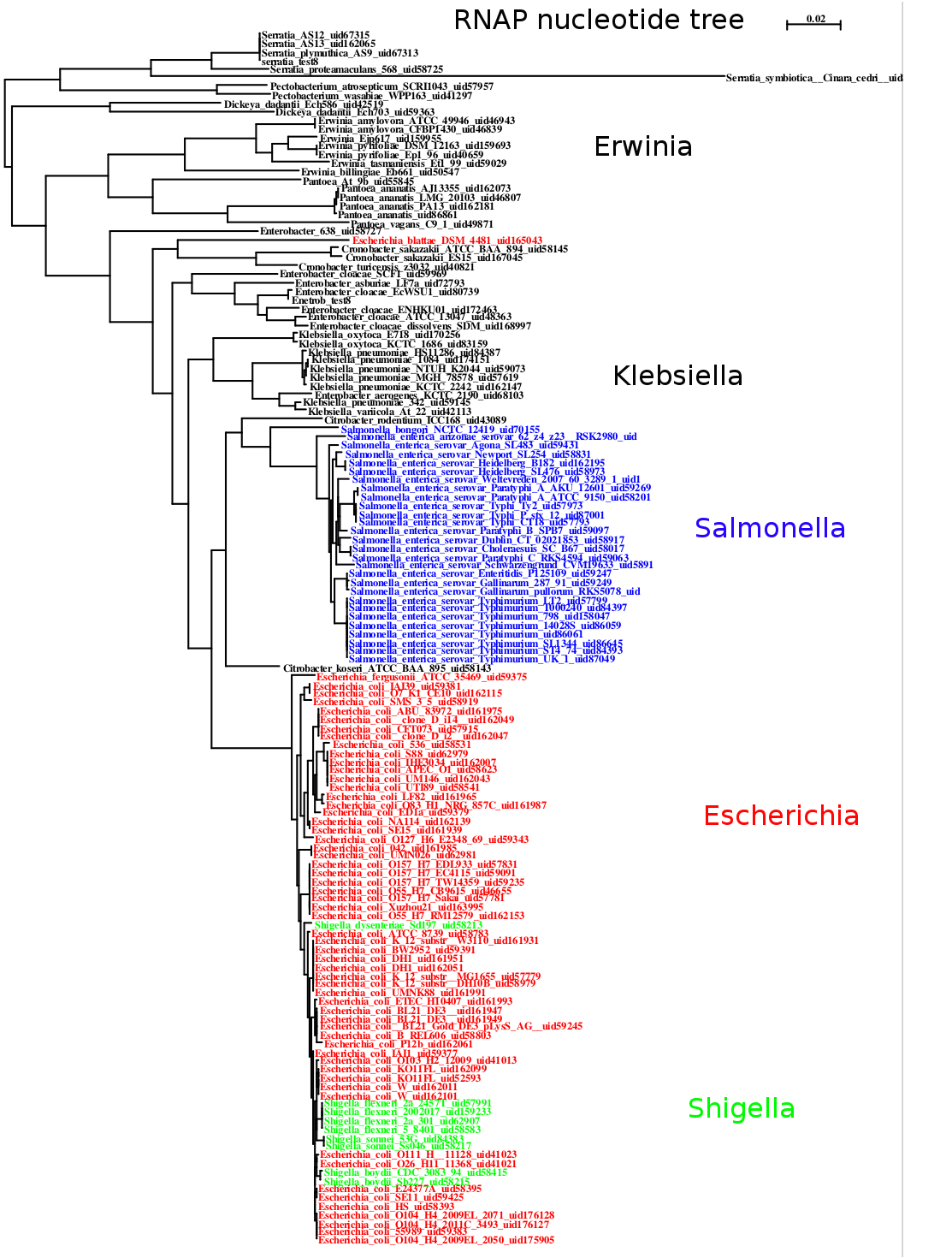
Maximum likelihood tree of near-neighbors of E. coli computed with PhyML from a nucleotide alignment of the beta and beta prime subunits of the RNA Polymerase genes.
The sequence alignments to make this type of tree are available for
E.coli.